When we think of pathogens, our minds jump to evil little microorganisms — be it viruses, bacteria, or parasites — that infect and wreak havoc on us from the outside. But we hardly think that such pathogens can originate within our own bodies and that they do not even have to be alive. Deadly prion diseases, for example, are caused by proteins, the very building blocks that are essential to our survival.
Prion diseases, caused by abnormal prion proteins, are rare and fatal neurological diseases that can be genetically inherited, emerge sporadically, or spread via transmission methods like blood transfusion and ingestion. The best-known of these is bovine spongiform encephalopathy, also known as mad cow disease. Spongiform encephalopathies are another name for prion diseases due to one of their hallmark symptoms, vacuolation, in which the brain takes on a sponge-like appearance when vacuoles, or holes, start to form. But what really are protein prions, how can they cause such drastic damage, and how are these diseases treated?
To answer these questions, it is important to look at a protein called PrPC, expressed in the brain and lymphatic system. Protein folding is crucial to the functionality of our cells, and when PrPC is misfolded into the infectious PrPSc, also known as the “prion,” it forms large clusters that build up in the brain and damage cells and tissues. However, it is neither the presence of PrPC nor PrPSc alone that is responsible for disease pathology. Rather, it is the conversion of PrPC to PrPSc that is so toxic.
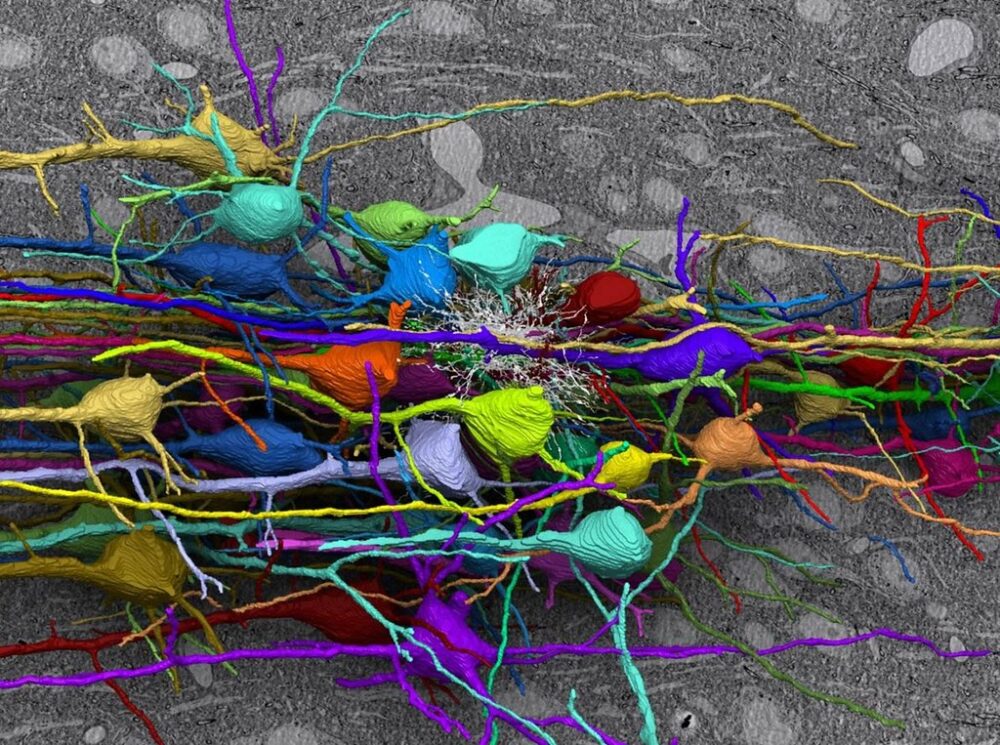
The reason behind this still remains a mystery but looking into the progression of prion diseases could offer some insights. Prion diseases begin with synaptic damage. Synapses are the gaps between neurons that allow them to communicate, and PrPSc aggregates hijack this machinery by accumulating in these spaces and interacting with synaptic proteins, interrupting neurotransmission. Following synaptic damage is dendritic loss. Dendrites are structures on neurons that receive signals from other neurons and are responsible for determining whether a neuron fires a signal. However, the presence of prions crumbles the actin cytoskeleton supporting dendrites and causes their spines to swiftly retract. While this is happening, PrPSc buildup also puts remarkable stress on the endoplasmic reticulum (ER), the cellular structure responsible for modifying proteins and relaying them to different parts of the cell. Because of this, the ER eventually activates the unfolded protein response, a defense strategy against the buildup of misfolded proteins that includes moving them out to the cytosol for degradation, activating ER chaperones, and reducing the amount of translation that takes place during protein synthesis. The brain’s immune response is also activated, leading to the next stage of the disease: brain inflammation.
The immune response of the brain is regulated by microglia, which are cells that digest pathogens. Microglia consume PrPSc deposits, which slows symptoms associated with their buildup. However, when the brain’s tools are forced to go into overdrive, they can be weaponized against us, and the stress produced by hyperactive microglia exacerbates neurotoxicity and accelerates neurodegeneration. It is also theorized that the activation of microglia, along with another type of brain cell, astrocytes, could actually drive pathology, as inflammation-related genes are upregulated alongside the earliest changes in the brain.
The next stage of disease, large-scale neuronal loss, is what leads to rapid deterioration and the poor prognosis of prion diseases. One way in which this takes place is vacuolation; although the mechanisms behind it are unclear, it is speculated that it could be caused by increased membrane permeability and water content within neurons or by autophagy, a process that digests cell organelles and misfolded proteins. Another process involved in neuronal death is apoptosis, a self-destructive mechanism driven by ER stress. ER stress also causes calcium to be released from the ER into the cytoplasm. Maintaining calcium concentrations is crucial for neuronal signaling, and the imbalance caused by this aberrant flux induces neuronal death.
“There is currently no cure for prion diseases, but there may be a way to prevent PrPC misfolding and prion replication”
There is currently no cure for prion diseases, but there may be a way to prevent PrPC misfolding and prion replication. Researchers found that synthetic compounds called antisense nucleotides can stop the production of PrPC, the normal protein — the disappearance of which has actually reversed symptoms of prion disease in mice. Additionally, short synthetic fragments of prion protein can stop the conversion of PrPC to PrPSc, but this only works during early stages of the disease. Future therapeutic strategies could involve targeting antibodies toward the N-terminus of the prion protein. Upon the presence of PrPSc, the N-terminal domain of the protein disconnects from the C-terminal domain, binding to the cell surface and initiating toxic effects. There is still much to learn and many exciting discoveries on the horizon, but one thing remains clear: The world of microbiology truly is a complex and surprising one.
Related posts:
 Secrets of the endurance athlete and how to become one
Secrets of the endurance athlete and how to become one
 When monsters under the bed come to life
When monsters under the bed come to life
 Genetic interventions in Alzheimer’s disease: A promising future
Genetic interventions in Alzheimer’s disease: A promising future
 Dead yet conscious: Some CPR survivors recall events during heart failure
Dead yet conscious: Some CPR survivors recall events during heart failure
 To die or not to die
To die or not to die
 The rise of Candida auris, a multidrug-resistant fungi
The rise of Candida auris, a multidrug-resistant fungi